View NGS data
GFF track
GFF file is a \t-delimited, 9-column plain text file. It mainly describes the interval information of various features on the genome, including chromosomes, genes, transcripts etc.
NC_000001.11 BestRefSeq pseudogene 11874 14409 . + . ID=gene0;Dbxref=GeneID:100287102,HGNC:HGNC:37102;Name=DDX11L1;description=DEAD/H-box helicase 11 like 1;gbkey=Gene;gene=DDX11L1;gene_biotype=transcribed_pseudogene;pseudo=true
NC_000001.11 BestRefSeq transcript 11874 14409 . + . ID=rna0;Parent=gene0;Dbxref=GeneID:100287102,Genbank:NR_046018.2,HGNC:HGNC:37102;Name=NR_046018.2;gbkey=misc_RNA;gene=DDX11L1;product=DEAD/H-box helicase 11 like 1;transcript_id=NR_046018.2
NC_000001.11 BestRefSeq exon 11874 12227 . + . ID=id1;Parent=rna0;Dbxref=GeneID:100287102,Genbank:NR_046018.2,HGNC:HGNC:37102;gbkey=misc_RNA;gene=DDX11L1;product=DEAD/H-box helicase 11 like 1;transcript_id=NR_046018.2
NC_000001.11 BestRefSeq exon 12613 12721 . + . ID=id2;Parent=rna0;Dbxref=GeneID:100287102,Genbank:NR_046018.2,HGNC:HGNC:37102;gbkey=misc_RNA;gene=DDX11L1;product=DEAD/H-box helicase 11 like 1;transcript_id=NR_046018.2
NC_000001.11 BestRefSeq exon 13221 14409 . + . ID=id3;Parent=rna0;Dbxref=GeneID:100287102,Genbank:NR_046018.2,HGNC:HGNC:37102;gbkey=misc_RNA;gene=DDX11L1;product=DEAD/H-box helicase 11 like 1;transcript_id=NR_046018.2
The following figure of SGS show genes and their corresponding transcripts structures in group. 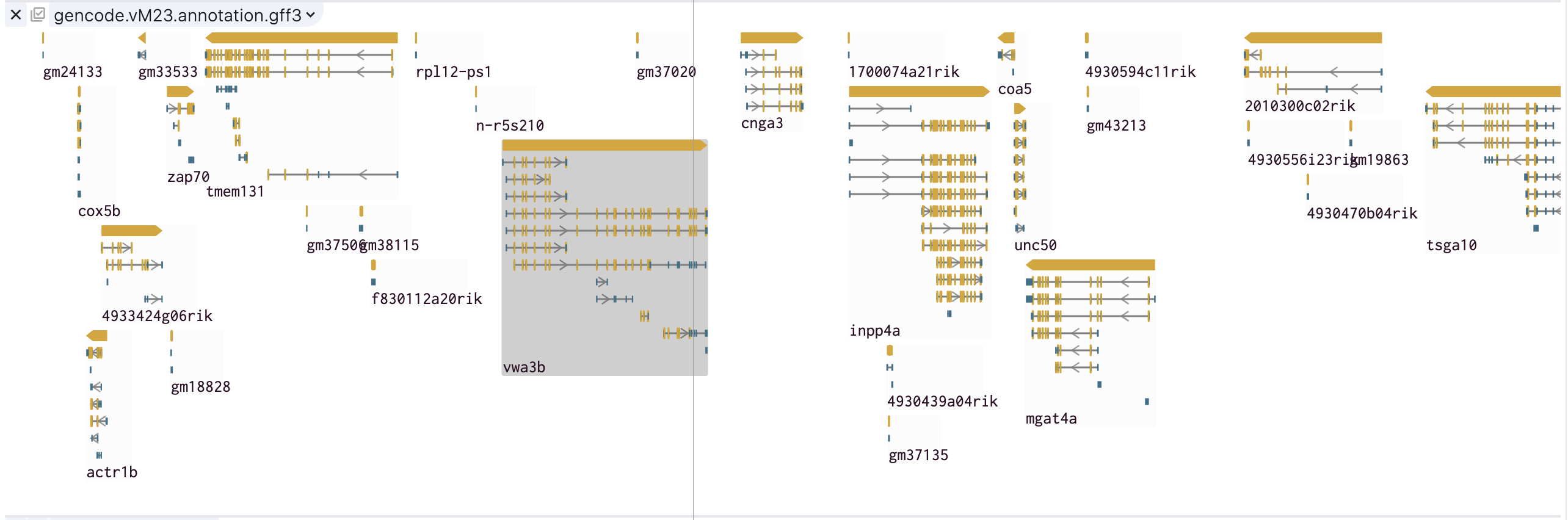
Users can adjust the style of GFF feature in track theme to view their interesting structures such as CDS, EXON, UTR. 
VCF track
VCF is the abbreviation of Variant Call Format(UCSC vcf format), which is a text file specially used to store gene sequence mutation information, including single base mutation SNV, single nucleotide polymorphism SNP, InDel, copy number variation CNV and structure Variant SV, etc. The beginning of the VCF file is the overall comment information, starting with ##, each line of the vcf body is a record, the fixed column has 9 columns, and the tenth column after that, they are:
| Column | Information |
|---|---|
| First column | CHROM: chromosome number; |
| Second column | POS: position on chromosome; |
| Third column | ID: mutation name; |
| Fourth column | REF: reference genome base type; |
| Fifth column | ALT: variant base type; |
| Sixth column | QUAL: the quality value of variant detection, the higher the more reliable |
| Seventh column | FILTER: the column that marks the filtering result; the one that passes the quality control filtering standard is marked as 'PASS'; |
| Eighth column | INFO: additional information column; |
| Ninth column | FORMAT: the description column of the following information; |
| Tenth column | sample information: GT=genotype, AD=number of bases supported, DP=sum of sequencing depth, PL=probability of genotype after normalization, GQ=quality value of genotype called by PL; |
##fileformat=VCFv4.0
##fileDate=20090805
##source=myImputationProgramV3.1
##reference=1000GenomesPilot-NCBI36
##phasing=partial
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2 membership">
##FILTER=<ID=q10,Description="Quality below 10">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 NA00002 NA00003
20 14370 rs6054257 G A 29 PASS NS=3;DP=14;AF=0.5;DB;H2 GT:GQ:DP:HQ 0|0:48:1:51,51 1|0:48:8:51,51 1/1:43:5:.,.
20 17330 . T A 3 q10 NS=3;DP=11;AF=0.017 GT:GQ:DP:HQ 0|0:49:3:58,50 0|1:3:5:65,3 0/0:41:3
20 1110696 rs6040355 A G,T 67 PASS NS=2;DP=10;AF=0.333,0.667;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2 2/2:35:4
20 1230237 . T . 47 PASS NS=3;DP=13;AA=T GT:GQ:DP:HQ 0|0:54:7:56,60 0|0:48:4:51,51 0/0:61:2
20 1234567 microsat1 GTCT G,GTACT 50 PASS NS=3;DP=9;AA=G GT:GQ:DP 0/1:35:4 0/2:17:2 1/1:40:3
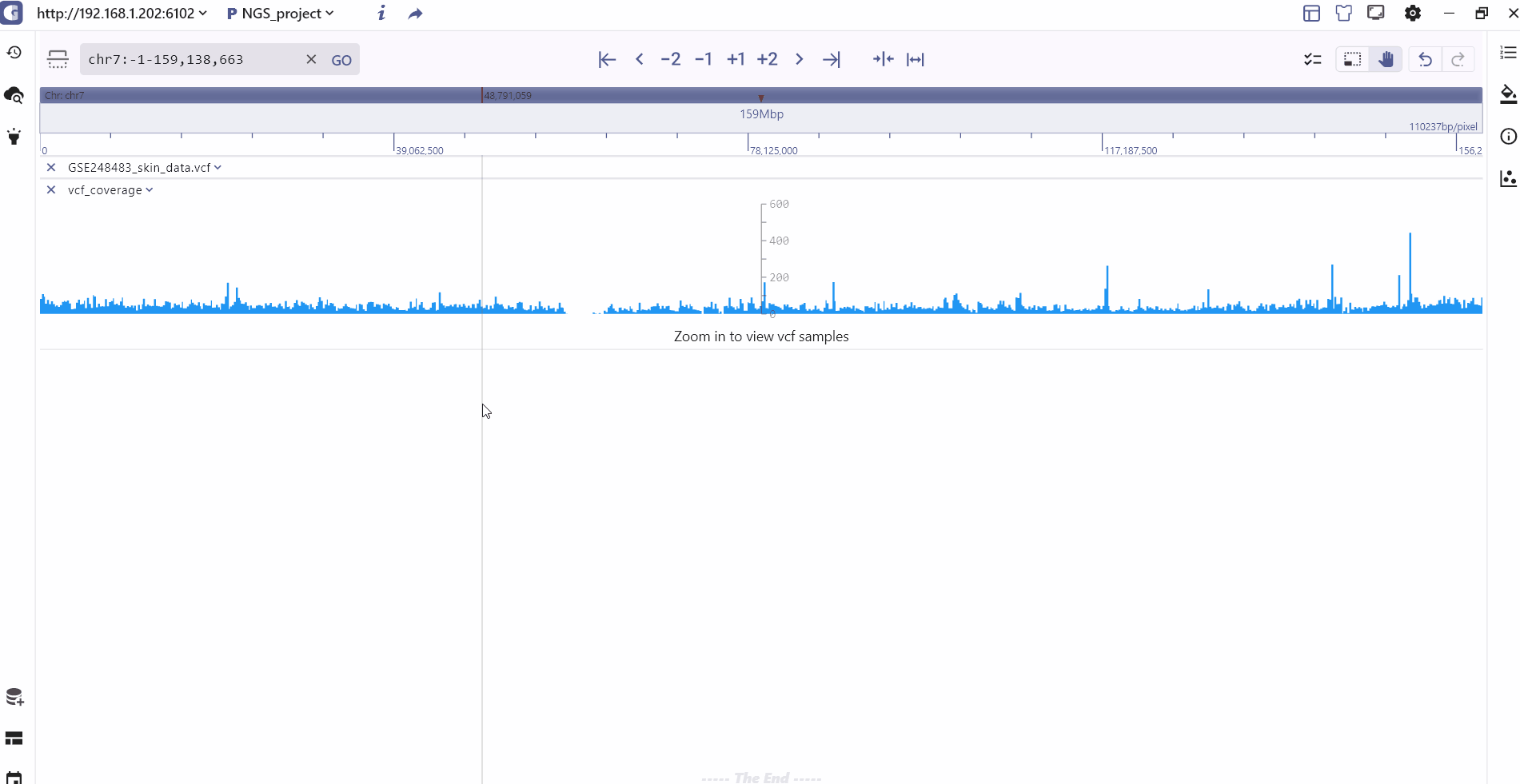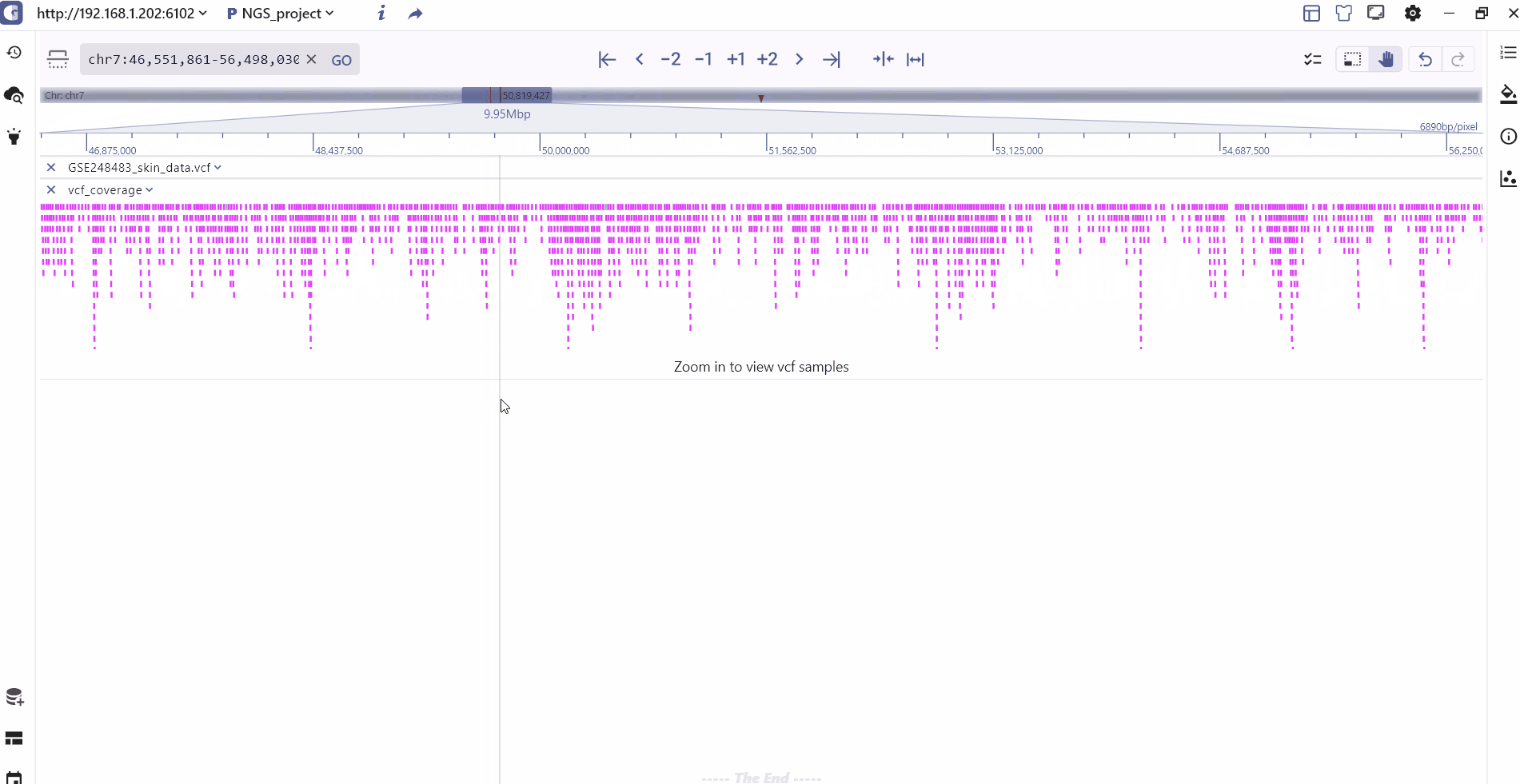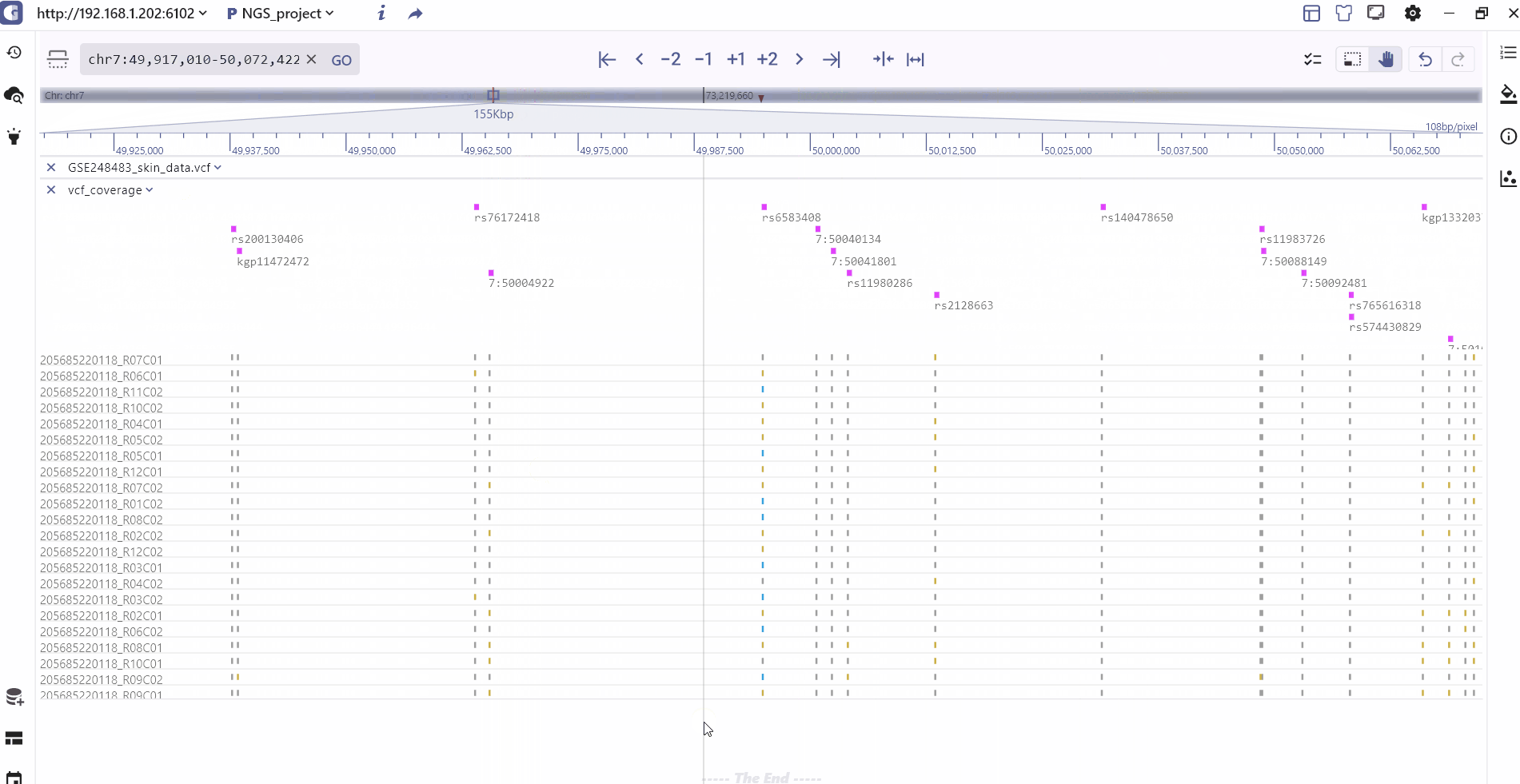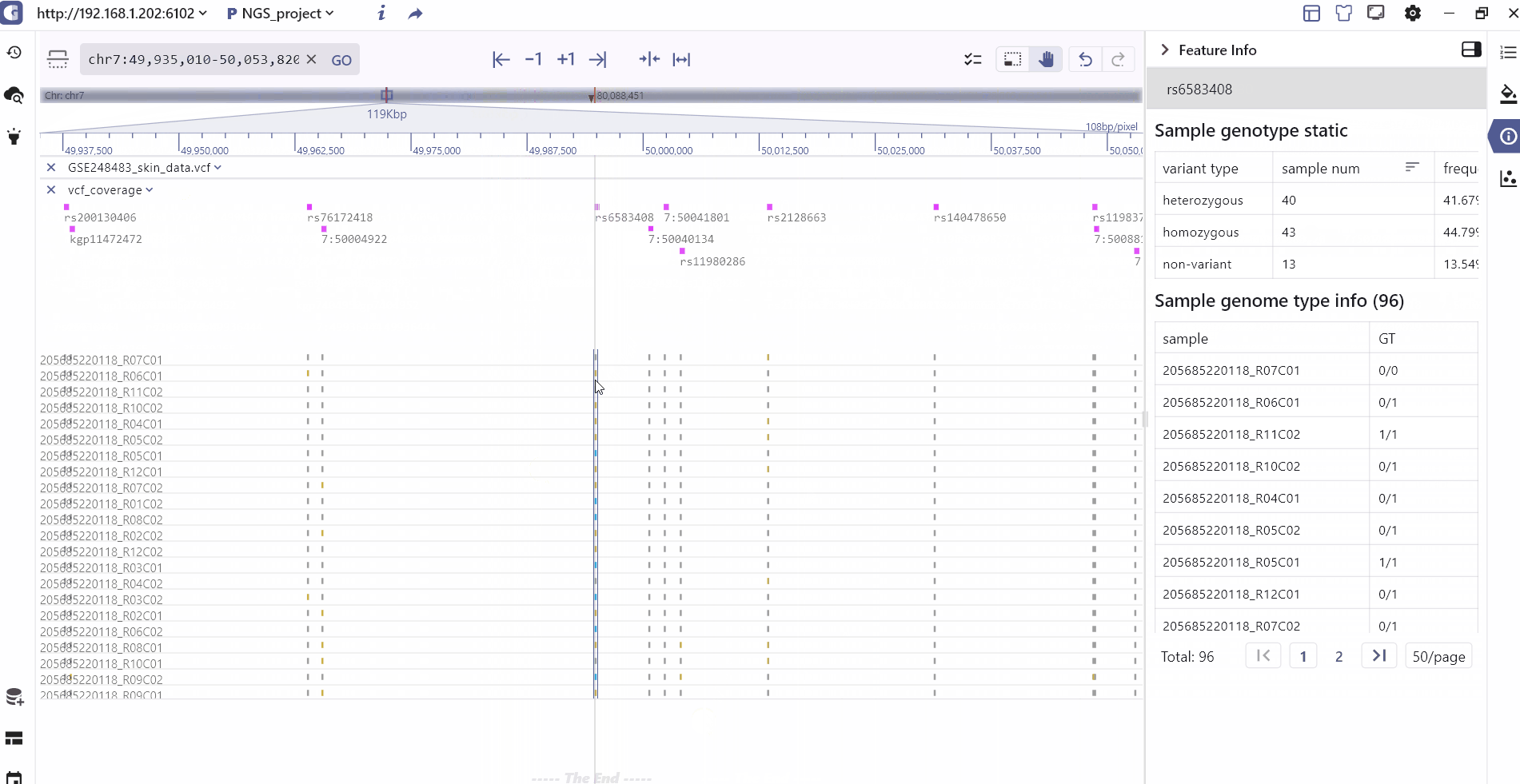
The VCF display in SGS includes the following information: It presents the variation position and type for each variant site, as well as the variation information for each sample. This includes statistical classification of sample variation information, genotype information (homozygous, heterozygous, non-variant), and specific details such as variant number and variant score.
BigWig Track
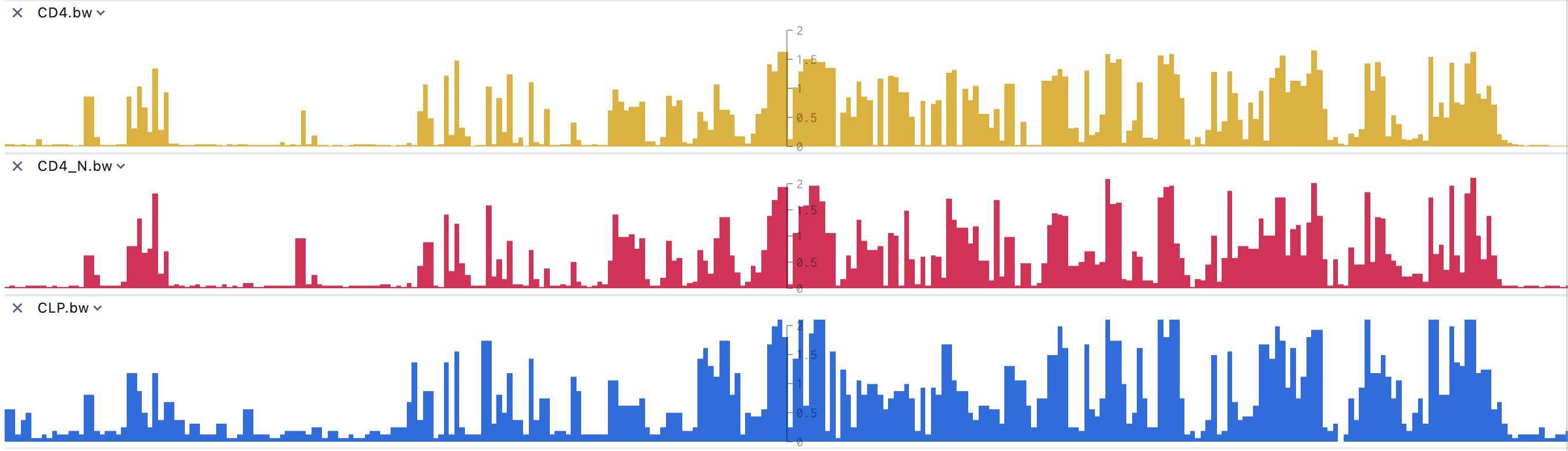
BigWig files are created from wiggle (wig) type files using the wigToBigWig program. BigWig files are indexed binary formats(UCSC bigwig format). The display performance of BigWig files is much faster than regular wiggle files when dealing with large datasets. Note: Wiggle data must be contiguous and consist of elements of equal size. If your data is sparse or contains elements of different sizes, use bedGraph format instead of wiggle format. If you have a very large bedGraph dataset, you can convert it to BigWig format using the bedGraphToBigWig program.
SGS displays dense, continuous data as graphs, with peaks representing scores at different locations. It offers various statistical modes such as max, min, mean, and sum, along with normalization methods like Linear, Pow.5, Log.
Bedgraph Track
Bedgraph is a \t-delimited, 4-column plain text file. It mainly used to records continuous data such as transcriptome or chip analysis data. The first column is , second column is , third column is and the last column is third column is .
chr19 49302000 49302300 -1.0
chr19 49302300 49302600 -0.75
chr19 49302600 49302900 -0.50
chr19 49302900 49303200 -0.25
chr19 49303200 49303500 0.0
chr19 49303500 49303800 0.25
chr19 49303800 49304100 0.50
chr19 49304100 49304400 0.75
chr19 49304400 49304700 1.00

Bed Track
A track for all bed-like files (with first column chromosome, second start and third end). If the other columns fit the requirement as defined in UCSC additional fields can be used. For example, the 5th and 9th column can be used to change the color of intervals, the 6th column indicate the strand. By default, intervals without strand are displayed as rectangle and for intervals with strand an arrow is added at the extremity (not included in the interval). In case of BED12 format, the introns are displayed into another color. But other styles are available.
chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7 127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7 127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
chr7 127475864 127477031 Neg1 0 - 127475864 127477031 0,0,255
chr7 127477031 127478198 Neg2 0 - 127477031 127478198 0,0,255
chr7 127478198 127479365 Neg3 0 - 127478198 127479365 0,0,255
chr7 127479365 127480532 Pos5 0 + 127479365 127480532 255,0,0
chr7 127480532 127481699 Neg4 0 - 127480532 127481699 0,0,255
At a larger zoom level, the SGS genome browser provides a variety of modes to display the statistical distribution of the number of bed fearures. 
At a smaller zoom level, it mainly displays the bed feature structure. 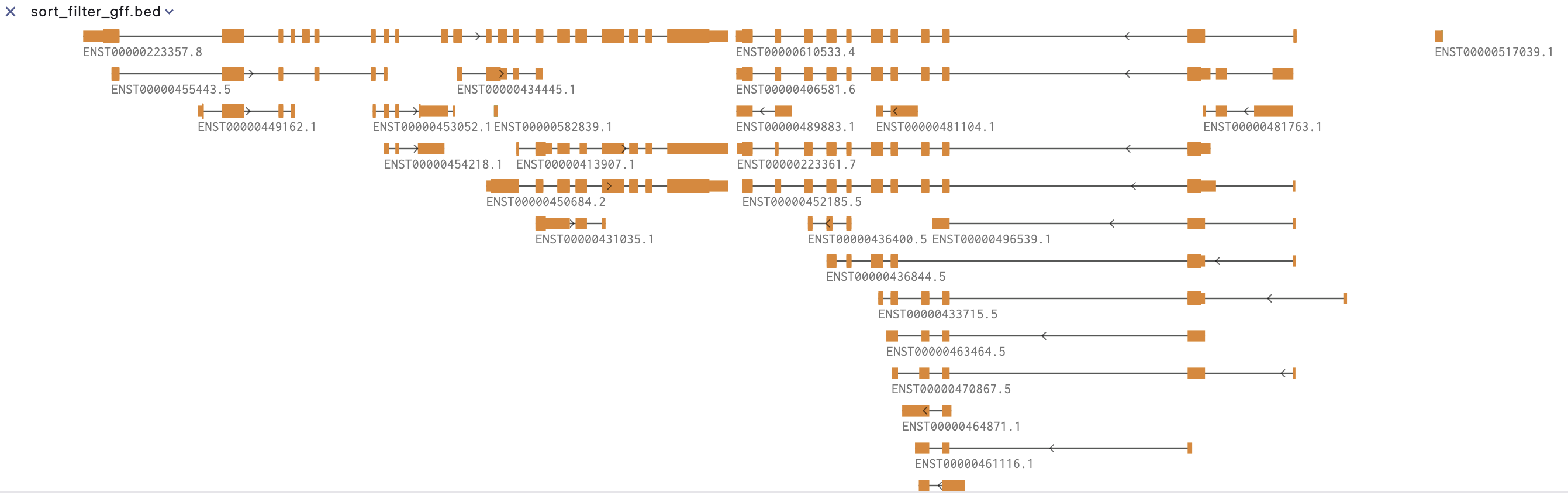
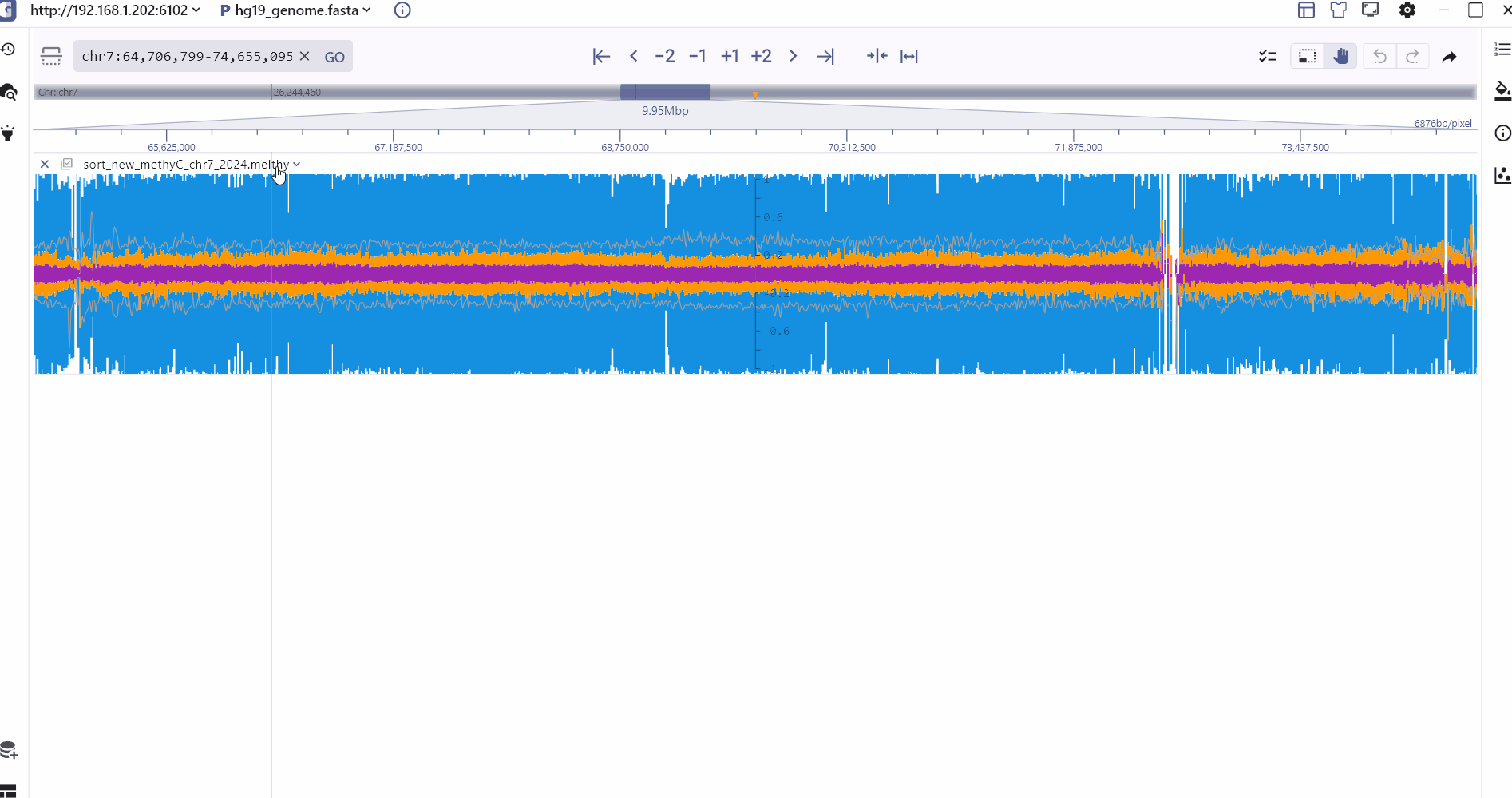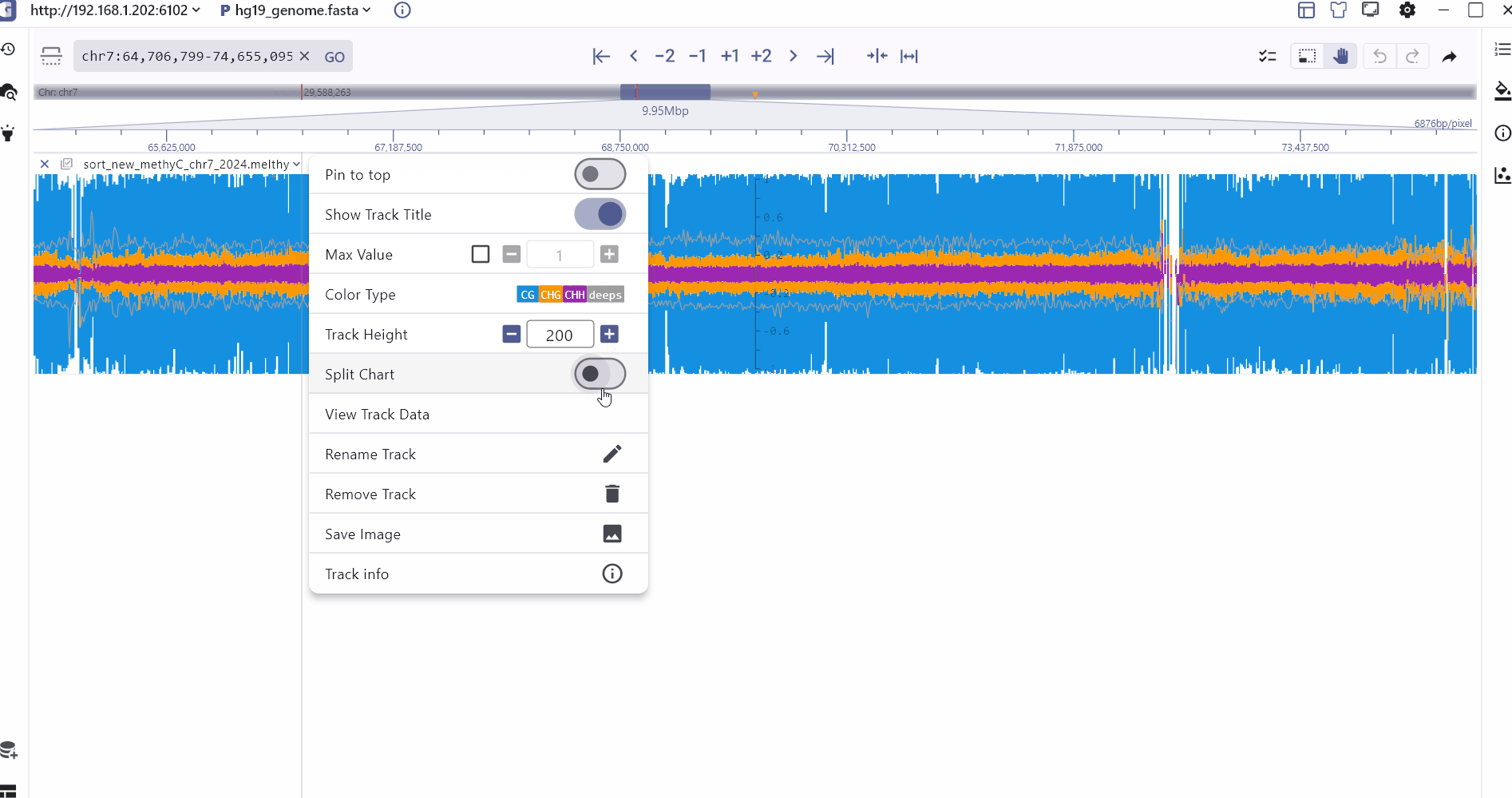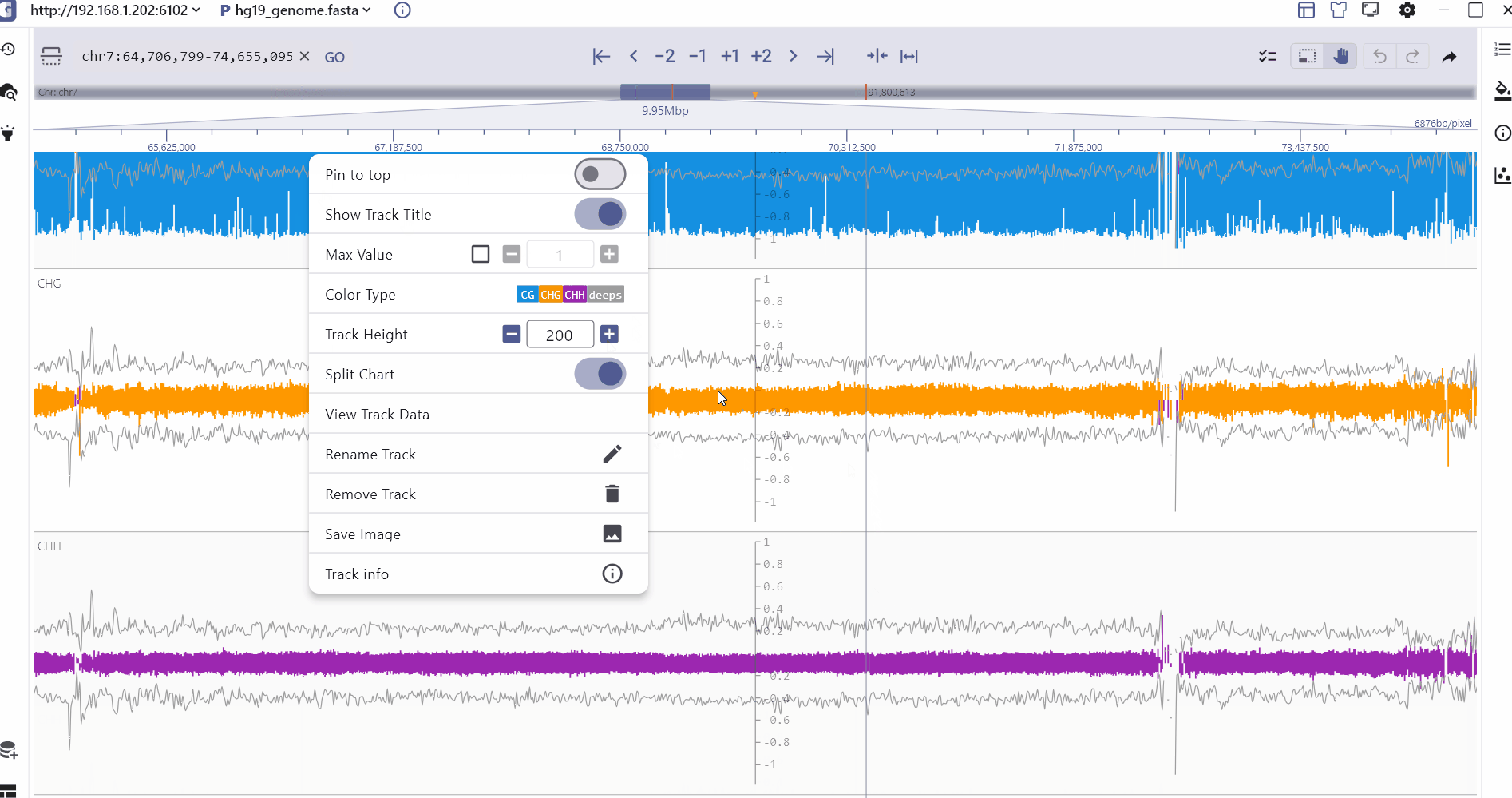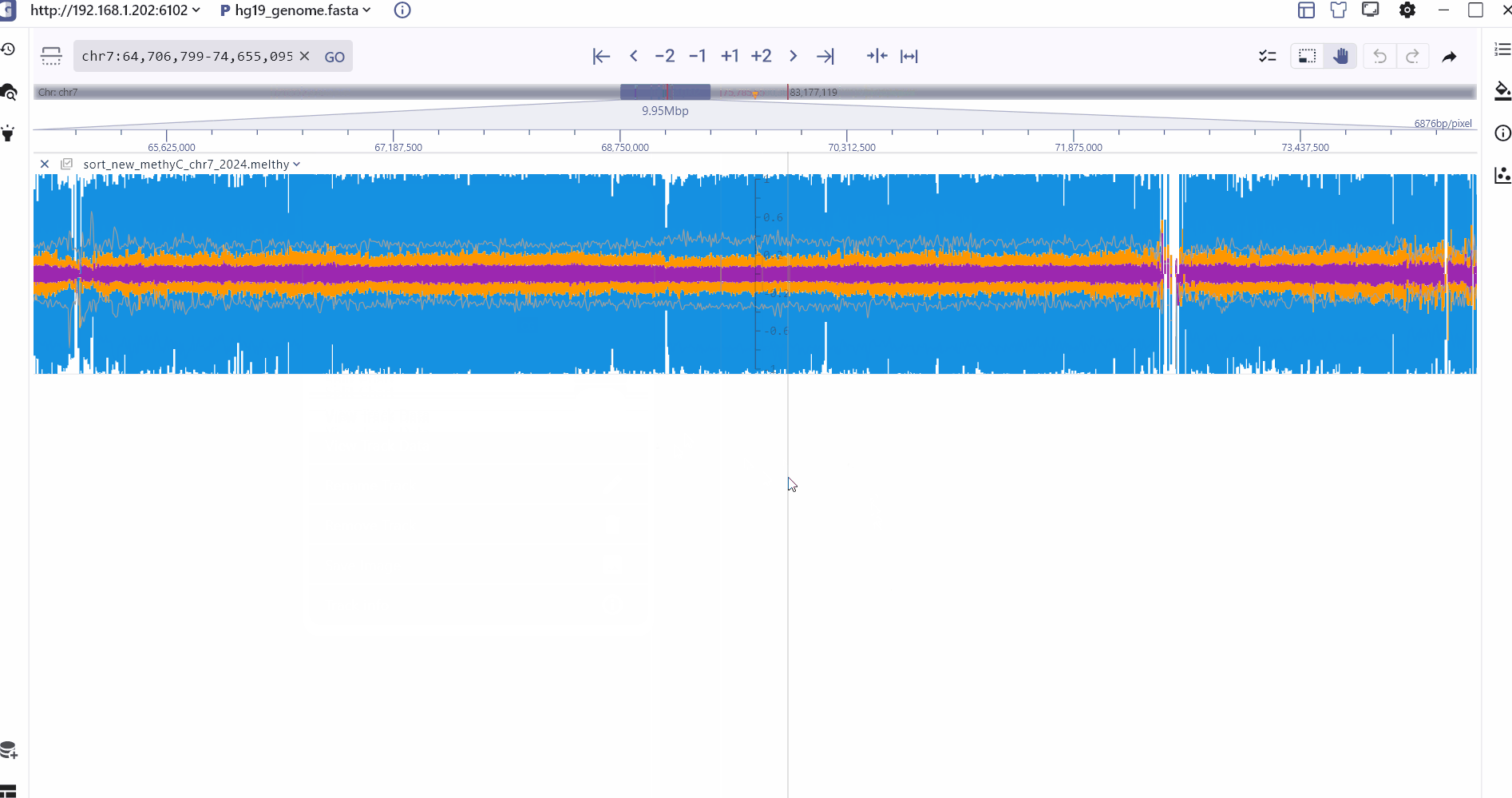
MethylC Track
The methylC file is a 7-column bed file that mainly records methylation at different genomic sites. From left to right, they are:, (0-based), (not included), (CG, CHH, CHG etc.), , , and .
chr1 10542 10543 CG 0.923 - 26
chr1 10556 10557 CHH 0.040 - 25
chr1 10562 10563 CG 0.941 + 17
chr1 10563 10564 CG 0.958 - 24
chr1 10564 10565 CHG 0.056 + 18
chr1 10566 10567 CHG 0.045 - 22
chr1 10570 10571 CG 0.870 + 23
chr1 10571 10572 CG 0.913 - 23
In order to better compare different methylation types, SGS displays CHG, CHH, CG and read depth information on the same track, and the specific methylation status of a genomic site can be viewed through mouse hover. In addition, the SGS genome browser also provides a split chart function to display the distribution of different methylation types separately.
Chromatin interaction Track
Hic
SGS mainly supports the visualization of .hic files, which mainly record the interaction information of different genome intervals. For specific hic data format, please refer to:hic format.
We provide two modes of heatmap and Arc to display the hic folder. Users can zoom to different levels to view the interaction at different resolutions, and the intensity of the interaction is displayed through the color depth. Users can view the two through the mouse hover. The interaction value of the interaction interval.

Longrange and biginteract
The longrange track is a bed format-like file type. It mainly consists of 4-6 columns, of which the first 4 columns are required and the last two columns are optional. From left to right are , (0-based), (not included), , (optional) and (optional). The target field mainly contains information such as the position of the target interaction interval and the interaction value. As an example, interval interacts with interval on a score of 2.
chr1 840086 840670 chr1:873317-874094,2 7837 +
chr1 856372 857075 chr1:873472-874198,2 7839 +
chr1 873317 874094 chr1:840086-840670,2 7838 -
chr1 873472 874198 chr1:856372-857075,2 7840 -
chr1 950622 951436 chr1:998665-999659,3 7841 +
chr1 974252 975234 chr11:518876-519376,2 71 .
chr1 974252 975234 chr11:518876-519376,2 69 .
chr1 998665 999659 chr1:950622-951436,3 7842 -
chr1 1056750 1057253 chr1:1165823-1166794,2 7843 +
chr1 1056981 1057755 chr1:1185448-1186693,3 7845 +
Biginteract
The interact format is available as a standalone plain text bed5+13 format. Biginteract is a binary indexed interact format. This format is useful for displaying functional element interactions such as SNP/gene interactions, and is also suitable for low-density chromatin interactions, such as ChIA-PET. For more details about this format, please check the UCSC biginteract format page.
SGS genome browser provides Arc mode, double mode and circle mode to display long-distance interaction.
Arc mode
Arc mainly shows the interaction of the same chromosome through arcs. 
Double mode
The double-layer mode is mainly viewed in the compare mode, which forms an interaction block through the interaction interval between the upper and lower chromosomes. The strength of the interaction through the intensity of the color. Users can zoom in/out the region by hovering the mouse wheel on a chromosome or switch the interaction range by drag left/right. 
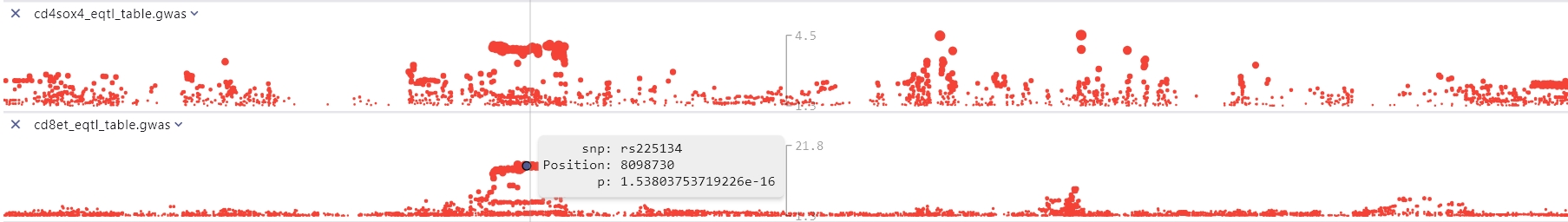
GWAS
A GWAS file is a space- or tab-delimited result file from genome-wide association study (GWAS) analysis. These files include PLINK result files containing integrated map information (i.e., chromosomal location for each association). File extensions for GWAS files are: .linear, .logistic, .assoc, .qassoc, .gwas GWAS file must contain a header line and four required columns (case-insensitive):
- CHR: chromosome (aliases chr, chromosome)
- BP: nucleotide location (aliases bp, pos, position)
- SNP: SNP identifier (aliases snp, rs, rsid, rsnum, id, marker, markername)
- P: p-value for the association (aliases p, pval, p-value, pvalue, p.value) Note: Columns can be in any order. Other columns besides the required ones are allowed and will be included in popup text. The p-value will be transformed to -log10 scale for plotting.
CHR BP RSID P-Value Gene ENSEMBL_ID Cell_Type A1 A2 FREQ_ONEK1K FREQ_HRC RHO FDR Genotyped ROUND
chr10 5855403 rs3736461 6.17087503323316e-12 GDI2 ENSG00000057608 Dendritic Cell G C 0.20625 0.184432 -0.218596159993423 0.000148710520344097 Imputed 1
chr10 71978144 rs7072230 2.11137407656829e-19 PPA1 ENSG00000180817 Dendritic Cell A G 0.33071 0.32045 0.283896081579831 0.000148710520344097 Imputed 1
chr10 71945039 rs7906276 0.000356168237078246 PPA1 ENSG00000180817 Dendritic Cell G A 0.80195 0.775516 0.114533481728928 0.0143271957821299 Imputed 2